

背景介绍
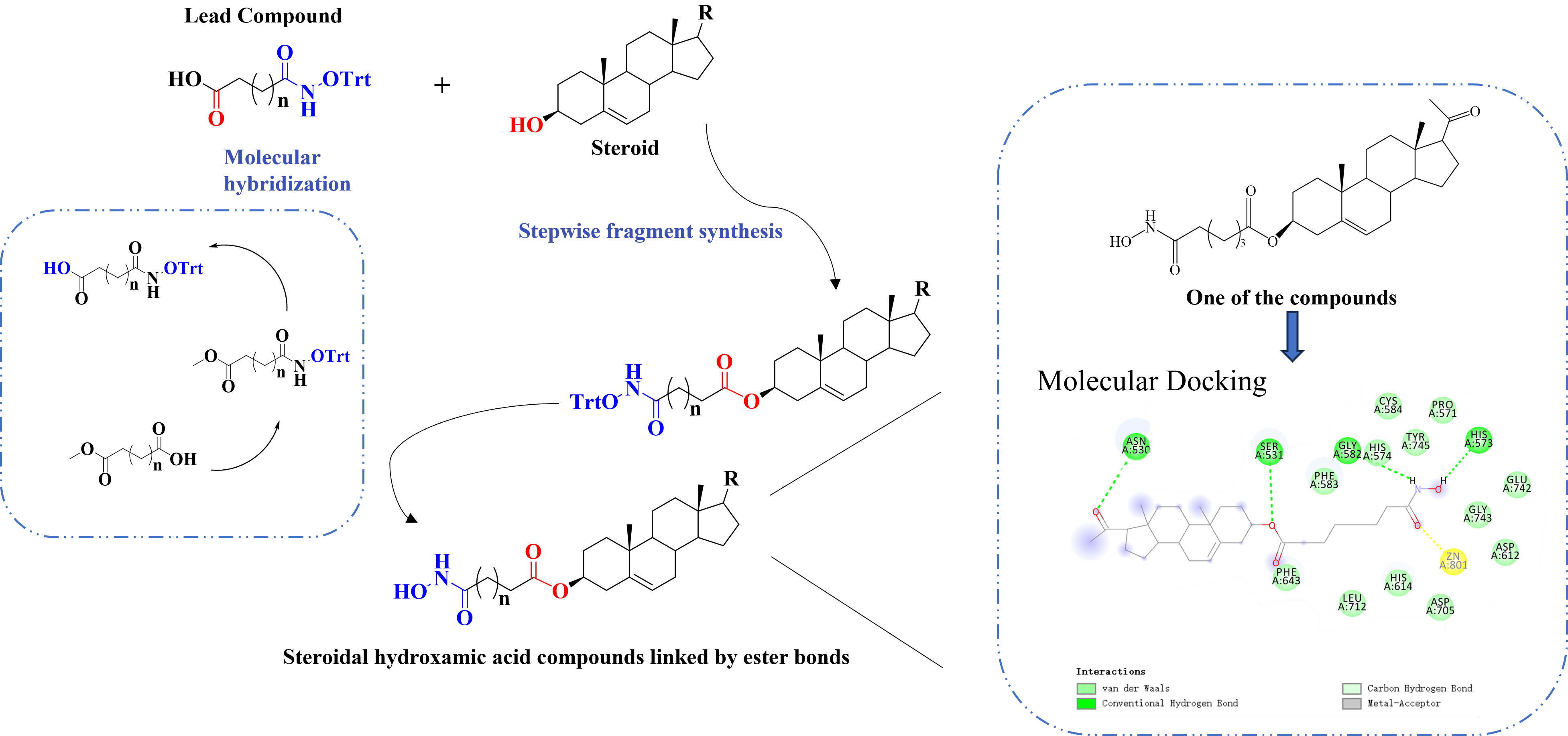
恶性肿瘤是全球重大健康挑战,现有疗法在疗效与毒性方面仍有局限。甾体化合物因其良好的生物相容性与多样生理活性,已成为抗肿瘤药物设计的重要骨架;而羟肟酸作为组蛋白去乙酰化酶(HDAC)抑制剂,在肿瘤治疗中显示出广阔前景。本研究基于“分子杂交”策略,将甾体与羟肟酸通过酯键缀合,构建了一系列结构新颖的甾体-羟肟酸缀合物,旨在开发兼具高活性与低毒性的抗肿瘤候选分子。
文章亮点
1. 结构创新与合成优化:首次通过酯键连接甾体与羟肟酸片段,设计并合成了系列新型缀合物。系统优化了合成条件,显著提高了关键中间体与目标产物的收率,为后续结构修饰与活性研究奠定了坚实基础;
2. 作用机制深入阐释:通过分子对接研究,揭示了化合物与HDAC酶的结合模式差异:部分依赖疏水相互作用增强结合亲和力,部分通过与锌离子结合发挥催化抑制潜力。首次明确了甾核结构与连接链长度对活性的协同调控关系,为理性药物设计提供了理论依据;
3. 类药性初步评估:通过计算机辅助ADMET分析,系统评估了候选化合物的吸收、分布、代谢与毒性特性。尽管存在细胞渗透性较低、蛋白结合率较高等挑战,部分化合物仍展现出中等药效开发潜力,为后续结构优化指明了方向。
内容介绍
1 实验部分
1.1 主要仪器与试剂
1.2 实验方法
2 结果与讨论
2.1 化学部分
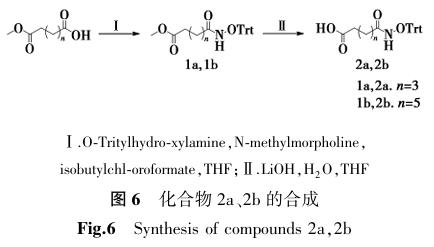
首先构建羟肟酸片段。以己二酸单乙酯和辛二酸单甲酯为原料,通过缩合反应及碱性水解法,合成氧代三苯甲基保护的胺基己酸、胺基辛酸及4-氧-(氧代三苯甲基-胺基)-苯甲酸中间体(图6)。
2.2 生物部分
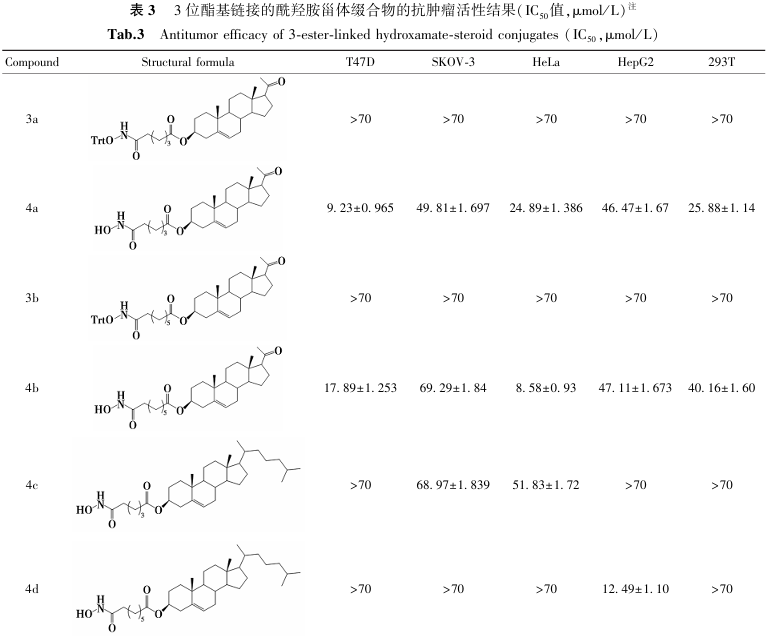
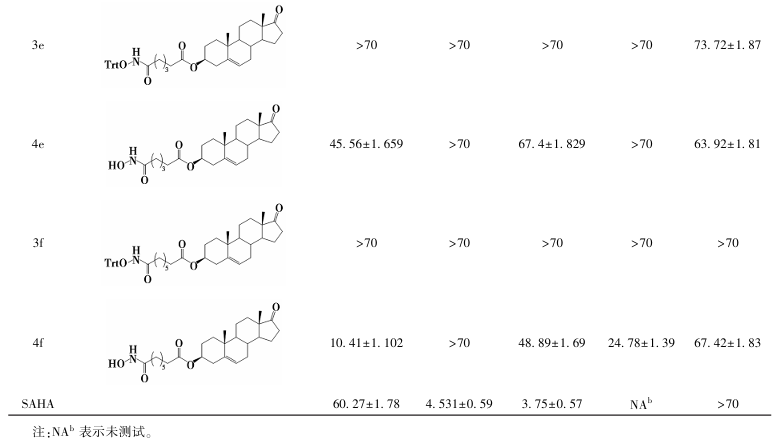
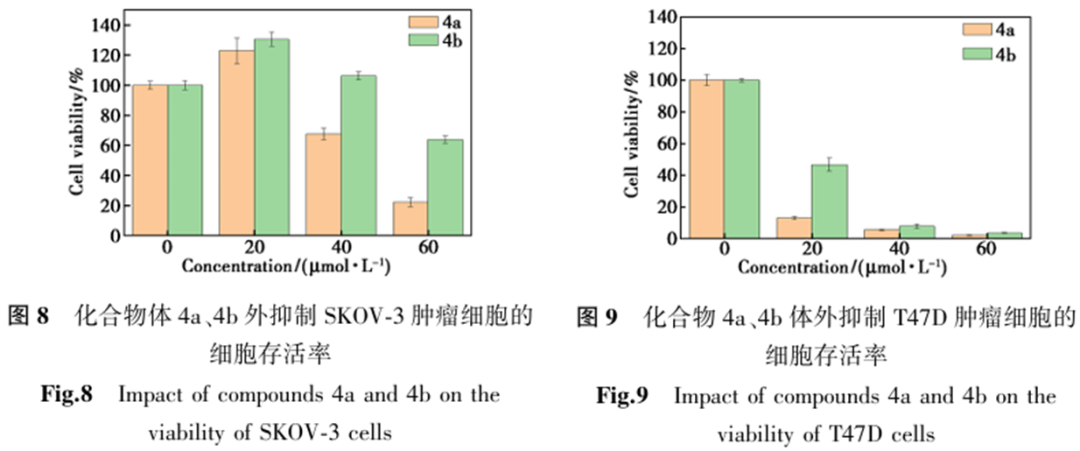
构效关系研究(表3、图8、图9)表明:1)胆甾烷结构支链的缀合物未显示肿瘤细胞抑制活性;2)对于孕烯醇酮缀合物,活性随支链碳原子数增加而减弱(化合物4a>4b);而对于去氢表雄酮缀合物,活性则随碳链增长而增强(化合物4e<4f);3)在相同取代基下,去氢表雄酮缀合物的细胞毒性优于孕烯醇酮缀合物;4)三苯甲基保护的酰羟胺前体无抗肿瘤活性,脱除保护基后的终产物均表现出良好活性,且部分化合物对T47D细胞的抑制活性显著优于其他测试细胞系。
2.3 分子模拟部分
2.3.1 分子对接(6V7A)
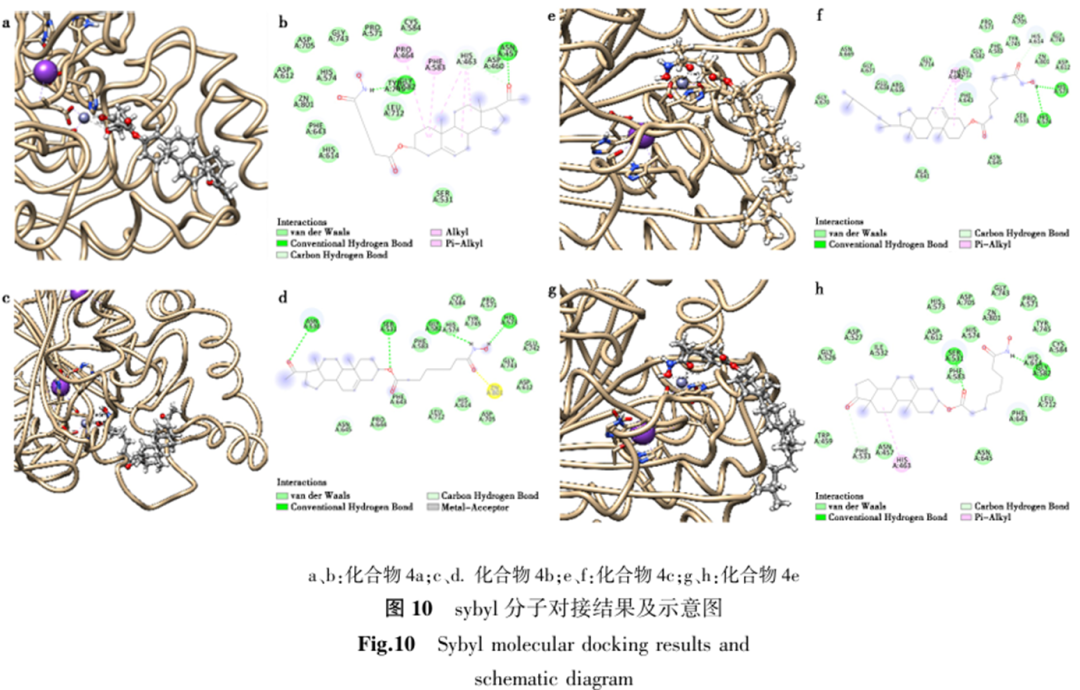
为深入探究目标化合物与SKOV-3和T47D肿瘤细胞的相互作用模式及结合亲和力,本课题组进一步针对HDAC 6(PDB ID:6V7A)进行了分子对接研究(图10)。
2.3.2 化合物4a、4b计算机辅助ADMET计算细胞毒性测试
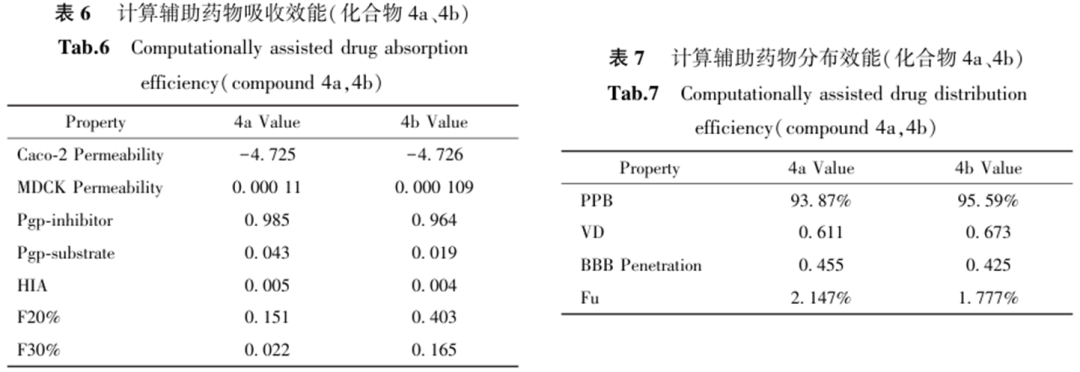
通过计算机辅助ADMET计算,对化合物4a和4b的吸收与分布性质进行了评估(表6、表7)。
3 结论
本研究采用分子杂交与分步片段合成策略,成功构建了一系列新型甾体-羟肟酸缀合物。通过系统优化,确立了以二氯甲烷为溶剂、DCC为缩合剂、DCC/DMAP物质的量之比1:1.5、反应温度35 ℃的最佳合成条件,关键中间体与目标产物收率良好,满足活性筛选需求。
活性研究表明:羟肟酸基团是必要药效团,孕烯醇酮与去氢表雄酮衍生物活性优于胆固醇衍生物。C4链利于T47D,C7链利于HeLa或HepG2,且与甾核结构协同作用。部分化合物在特定细胞系(如T47D)中活性优于SAHA,具备选择性抗肿瘤潜力。机制研究揭示,化合物4a与4b的作用机制存在差异:化合物4a主要通过疏水相互作用展现突出的结合亲和力,而化合物4b则凭借与锌离子的结合在催化抑制方面更具潜力。ADMET预测提示两者虽存在吸收与毒性方面的挑战,但仍具中等药效潜力,后续优化应聚焦于提高选择性,平衡效价与细胞毒性,进一步结构优化与深入研究。


