

背景介绍
丙酮酸及其酯类衍生物在食品、医药等领域广泛应用,但目前报道的检测方法多为纯品或单组分检测,不适合用于基质复杂的反应液中丙酮酸乙酯和丙酮酸的同时测定。在乳酸乙酯氧化法制备丙酮酸的连续生产过程中,需同时关注氧化工段与水解工段中中间体丙酮酸乙酯和产物丙酮酸的含量变化趋势,因此,需建立一种能同时测定氧化体系反应液中的丙酮酸和丙酮酸乙酯含量的分析方法。高效液相色谱法(HPLC)具有高分辨率、快速分析、高重复性等特点;抗坏血酸能有效淬灭氧化试剂;2,4-二硝基苯肼衍生(DNPH)可向丙酮酸与丙酮酸乙酯中引入强发光基团,提高检测的灵敏度与稳定性;衍生-HPLC法可实现基质复杂的反应液中中间体丙酮酸乙酯和产物丙酮酸的快速同时测定。
文章亮点
1.报道了一种专属性强、灵敏度高的衍生-HPLC分析方法,用于同时测定反应液中丙酮酸与丙酮酸乙酯的含量;
2.通过添加抗坏血酸淬灭反应液中的双氧水,有效防止双氧水破坏丙酮酸与衍生剂;
3.用DNPH作为衍生剂,引入强发光基团,提高了丙酮酸和丙酮酸乙酯的检测灵敏度与稳定性;
4.方法可应用于丙酮酸制备过程中的反应监控,对丙酮酸生产过程中产率和转化率的评估提供数据支持。
内容介绍
1 实验部分
1.1 主要仪器与试剂
1.2 实验方法
1.2.1 溶液配制
1.2.2 色谱条件分析
中谱红RD C18色谱柱(250 mm×4.6 mm×5 μm);柱温35 ℃;检测波长360 nm;进样量10 μL;流速1.0 mL/min;流动相A为乙腈,流动相B为0.1%磷酸水溶液;梯度洗脱程序:0~12 min,40% A+60% B→85% A+15% B;12~18 min,85% A+15% B→85% A+15% B;18~18.01 min,85% A+15% B→40% A+60% B,18.01~27 min,40% A+60% B。
1.2.3 样品的衍生化处理
精确称取0.4 g反应液,置于50 mL容量瓶中,加入溶剂溶解并稀释至刻度;移取1 mL上述溶液,置于50 mL容量瓶中,依次加入2 mL淬灭剂溶液、1 mL盐酸溶液与2.0 mL衍生剂溶液,于40 ℃水浴中衍生20 min,冷却至室温,再用溶剂稀释至刻度,摇匀,作为样品溶液。
2 结果与讨论
2.1 前处理条件的考察
2.1.1 衍生剂的选择与VC的添加
2.1.2 淬灭剂、盐酸与衍生剂加入量
2.1.3 衍生温度和时间
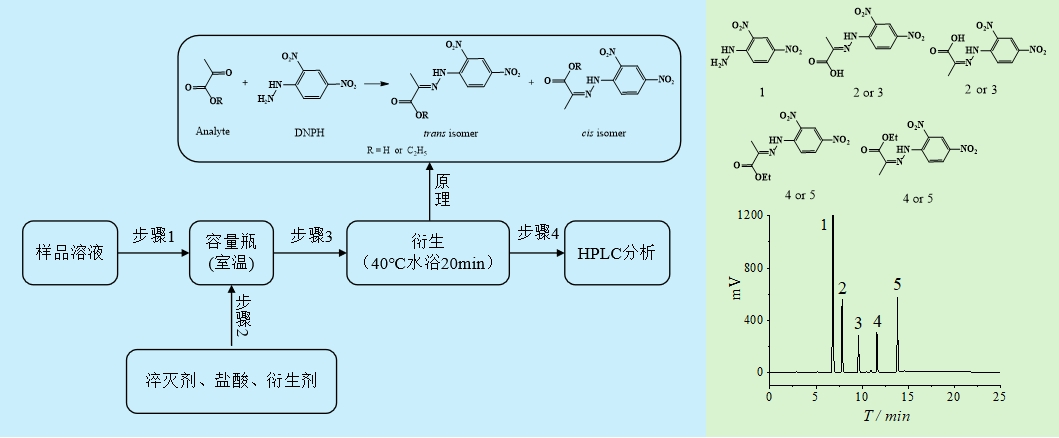
试验考察衍生温度对模拟液样品中丙酮酸与丙酮酸乙酯的测定的影响。当衍生时间为20 min时,衍生温度分别为20、25、30、35、40、45、50、55与60 ℃时,丙酮酸与丙酮酸乙酯的回收率测定结果见图5。
试验考察衍生时间对模拟液中丙酮酸与丙酮酸乙酯测定的影响。当衍生温度为40 ℃时,衍生时间分别5、10、20、30与40 min,丙酮酸与丙酮酸乙酯回收率结果见图6。
2.2 色谱条件的优化
2.2.1 检测波长的选择
根据DNPH性质,以及丙酮酸与丙酮酸乙酯衍生物的化合物性质,选择360 nm作为检测波长,有效避开试样中杂质与空白溶剂峰干扰。
2.2.2 流动相和检测条件选择
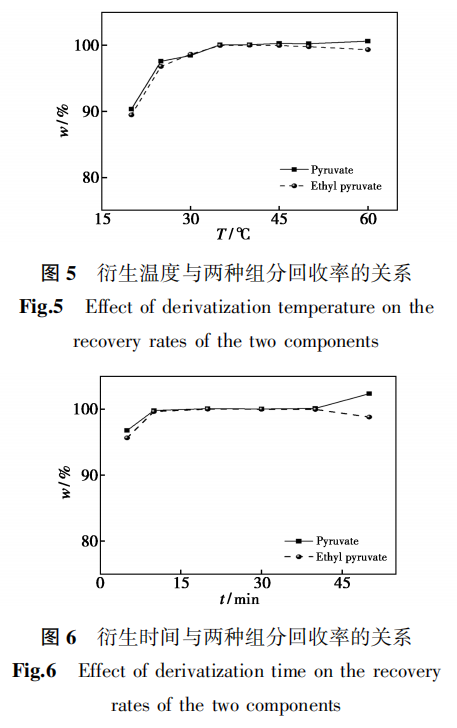
丙酮酸和丙酮酸乙酯衍生物为两性化合物,其电离形式随着溶液pH变化而变化,因此,考察了不同pH值流动相对丙酮酸和丙酮酸乙酯分离效果的影响。采用水+乙腈作为洗脱体系时,在C18上进行洗脱,丙酮酸乙酯分为两个色谱峰,峰型均较好,而丙酮酸衍生物的色谱峰峰形较差(图7a);采用0.1%磷酸溶液+乙腈作为洗脱体系时,丙酮酸乙酯分为两个色谱峰,丙酮酸为两个色谱峰,且各色谱峰峰形较好、分离度均大于2.0(图7b)。试验0.1%磷酸溶液+乙腈为流动相,C18柱为色谱柱。
2.3 色谱行为
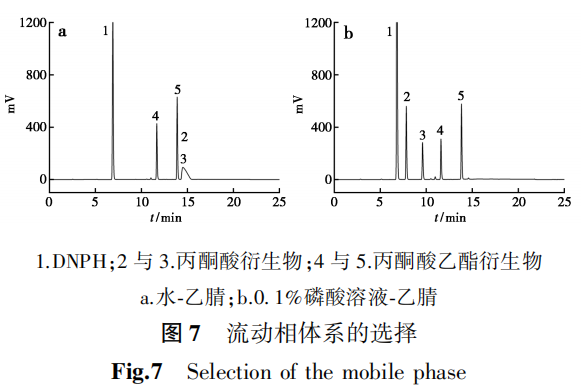
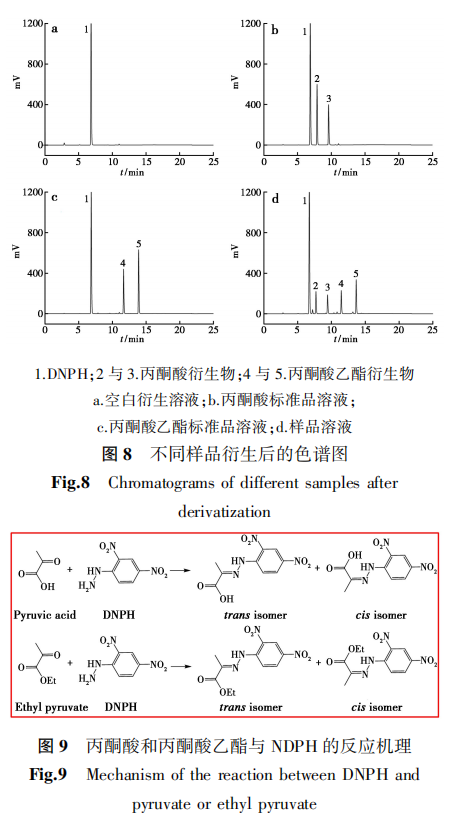
分别量取空白衍生溶液、丙酮酸标准品溶液、丙酮酸乙酯标准品溶液、样品溶液,按照1.2.2色谱分析条件进行测定,试验结果见图8。由于丙酮酸和丙酮酸乙酯与DNPH生成的腙类衍生物存在顺反异构体,在检测时会分别产生两个邻近的色谱峰,反应机理见图9。
2.4 方法学考察
3 结论
本研究建立了衍生-HPLC相结合同时测定反应液中丙酮酸与丙酮酸乙酯的分析方法。通过优化前处理条件与色谱条件,以DNPH为衍生剂,以VC为淬灭剂,考察衍生温度与衍生时间等因素及检测波长与流动相的pH值等因素,确认了丙酮酸与丙酮酸乙酯检测的分析方法。丙酮酸与丙酮酸乙酯衍生物的线性关系、精密度、准确度与溶液稳定性均符合要求,表明该方法专属性强、灵敏度高、操作简便,方法适用性强,可应用于乳酸乙酯制备丙酮酸反应过程中的反应监控,并为各工段丙酮酸和丙酮酸乙酯产率和转化率的评估提供数据支持,对丙酮酸制备工艺的反应监控和产率情况评估具有指导意义。
通讯作者介绍

岳涛
个人简介
主要研究方向
精细化学品绿色合成技术的开发、检测及产学研用一体化研究。


