




【试剂前沿】上海交大张万斌院士团队最新JACS!模块化合成新工具

文章原文:【试剂前沿】上海交大张万斌院士团队最新JACS!模块化合成新工具
模块化合成新工具:手性双环咪唑催化助力手性膦氧化物的高效构建
近日,上海交通大学张万斌教授课题组在手性膦氧化物的合成中取得了新的进展:基于手性双环咪唑催化剂的双功能催化作用,通过对膦酰二氯的高效去对称化反应一步构建磷手性中心,并结合“一锅法”策略在第二步中进行立体特异性地SN2亲核取代反应,实现了手性膦氧化物的多样化合成。相关成果发表在J. Am. Chem. Soc.上。
正文
P-手性有机膦氧化合物,含有一个或两个C-P键,是一类具有重要价值的结构,在药物化学和有机合成中具有广泛应用。这些手性P(V)化合物广泛存在于许多天然产物、药物和生物活性分子中。在含有一个C-P键的化合物中,替诺福韦艾拉酚胺是治疗慢性乙型肝炎的一线药物;含有两个C-P键的化合物也可由其单C-P键对应物衍生而来,展现出多样化的生物活性。例如,Phostine表现出有效的抗增殖特性(图1)。
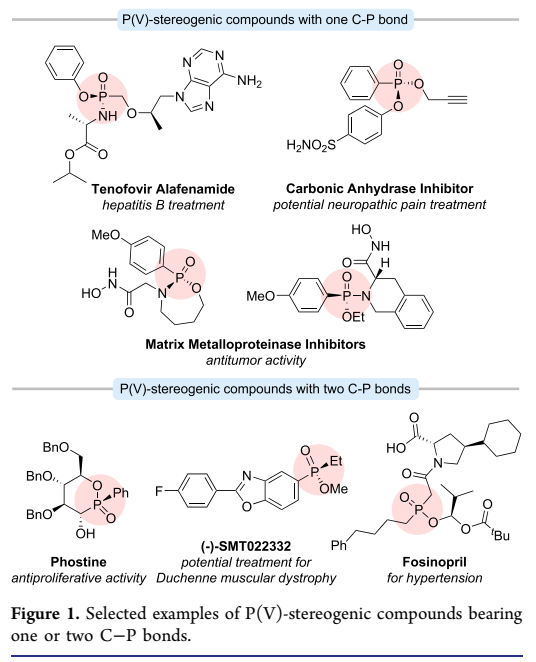
图1 含有C-P键的膦氧化物的重要性
由于其立体构型对活性具有重要影响,因此发展高效、高选择性合成P-手性中心的方法成为该领域的关键挑战。传统合成路径主要依赖外消旋体拆分与手性辅助基诱导策略,前者受限于50%的理论产率上限,后者则需引入并后续移除手性导向基团,步骤繁琐且原子经济性低。相比之下,不对称催化能够直接构筑P-手性中心,具有步骤简洁的优势,是当前该领域的研究重点。近年来,催化不对称去对称化策略在构建含C-P键的P(V)手性分子方面取得重要进展(图2A),Jacobsen团队(Science2022, 376, 1230−1236)、Dixon、Yamazaki(Nat. Chem.2023, 15, 714−721; Angew. Chem. Int. Ed. 2024, 63, e202400673)和Chi(Chem 2025, 11, 102586)等课题组先后开发了基于氢键给体、超强碱双功能氨基磷烷及N‑杂环卡宾等催化体系,为P‑手性化合物的合成开辟了新途径。这些工作与李光勋(J. Am. Chem. Soc. 2025, 147, 11010−11018)、何智涛(J. Am. Chem. Soc. 2025, 147, 13566−13576)、Dong (J. Am. Chem. Soc. 2025, 147, 21339−21346)及商明 (Angew. Chem. Int. Ed. 2025, 64, e202509807)等课题组的全杂原子取代的磷(V)手性中心的研究共同推动了该领域发展。另一方面,催化不对称磷酰化也是构建P-手性中心的重要策略(图2B)。然而,现有含C-P键手性膦氧化合物的不对称合成方法,在催化剂可获得性、反应普适性与对映选择性方面仍存在不足,亟需发展更为高效、通用且催化剂易于制备的新型催化体系。自2010年始,上海交通大学张万斌课题组基于“键角调控”策略开发了一系列具有双环咪唑结构的有机小分子催化剂。利用该类催化剂,他们报道了首例构建磷(V)手性的不对称磷酰化反应,并成功地实现了磷(V)手性抗新冠药物瑞德西韦的高效不对称合成(J. Am. Chem. Soc. 2010, 132, 15939-15941; Tetrahedron: Asymmetry 2012, 23, 329−332; Chem. Commun.2017, 53, 1381-1384; Angew. Chem. Int. Ed. 2020, 59, 20814-20819; Angew. Chem. Int. Ed. 2021, 60, 1641-1645; CCS Chem. 2023, 5, 361-371; ACS Catal. 2023, 13, 16300-16306)。基于此,最近该团队报道了一种手性双环咪唑双功能手性催化剂,能经由膦酰二氯去对称化及后续的立体专一性SN2亲核取代过程,从而一锅法地实现多种结构的手性膦氧化合物的合成(图2C)。该方法可以高效制备系列含有P-O/P-S/P-N键结构的P(V)-手性分子,且收率与对映选择性表现优异。此外,所得产物还可以进一步被醇解,生成的手性膦酸酯又可以多样化精准衍生,构筑新的P-S/P-C/P-C(Ar)键,为合成具有生物活性的高价值手性膦氧化合物提供了通用平台。
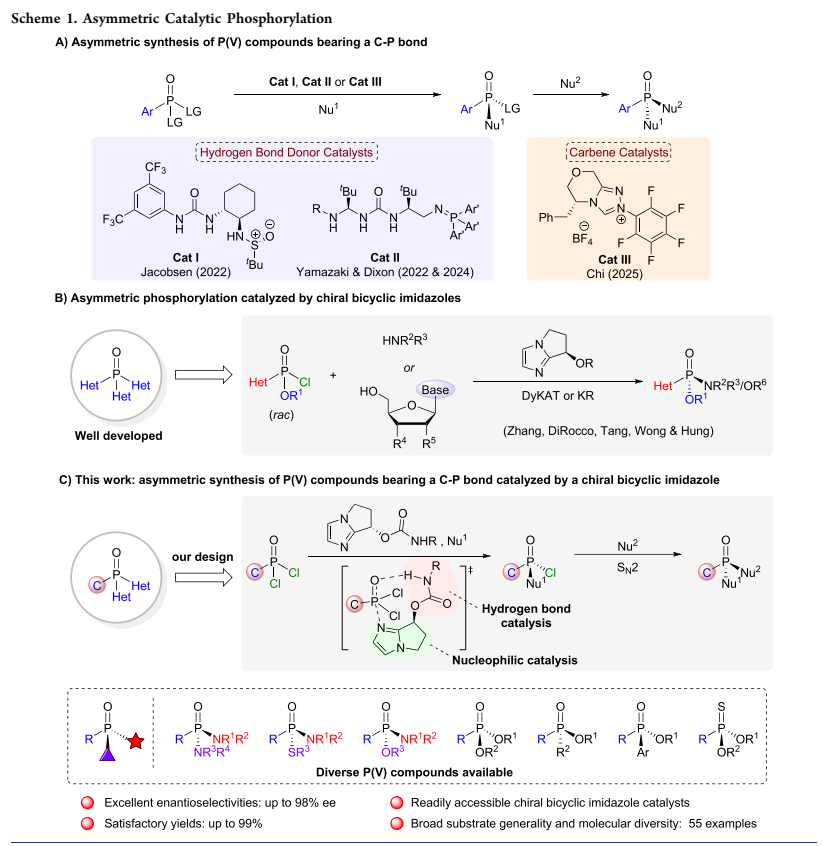
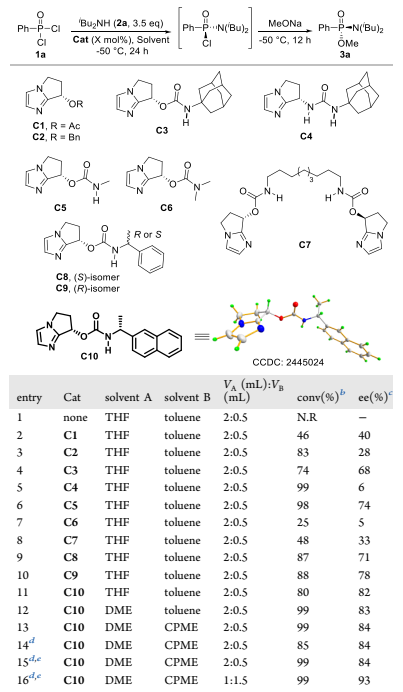
图 3 部分条件优化
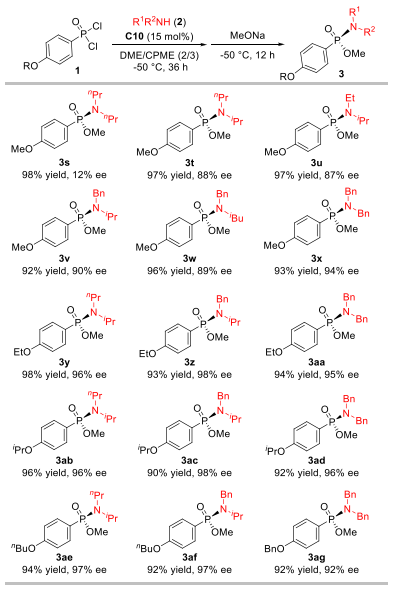
作者首先以市售的苯基膦酰二氯为模板底物,二异丁胺作亲核试剂与缚酸剂,并以四氢呋喃和甲苯分别作反应溶剂与底物溶剂,在-50 °C的低温条件下对催化剂进行了筛选和优化。实验结果表明,不添加催化剂时,反应无法进行。C1和C2催化剂在不对称C-酰化反应中展露出较好的效果,但在此条件下,该催化剂的立体控制效果有限(entry 2-3)。基于C3在瑞德西韦的不对称合成中的优异表现,研究团队尝试其催化该反应,获得74%转化率和68% ee(entry 4)。进一步结构修饰发现:将C3的氨基甲酸酯结构片段换为脲基(C4)可提高反应活性但损害对映选择性(entry 5);N–H键对高催化性能至关重要(C5/C6,entry 6-7)。随后筛选多种NH-取代催化剂,确定了最优催化剂C10表现出最高立体选择性(82% ee,entry 11)。随后进行溶剂以及反应时间等条件的优化,最终确定以1 mL DME为反应溶剂,1.5 mL CPME溶解底物,催化剂负载降至15 mol%,第一步反应36小时,随后使用甲醇钠淬灭可在高转化率下保持高对映选择性(entry 16)。
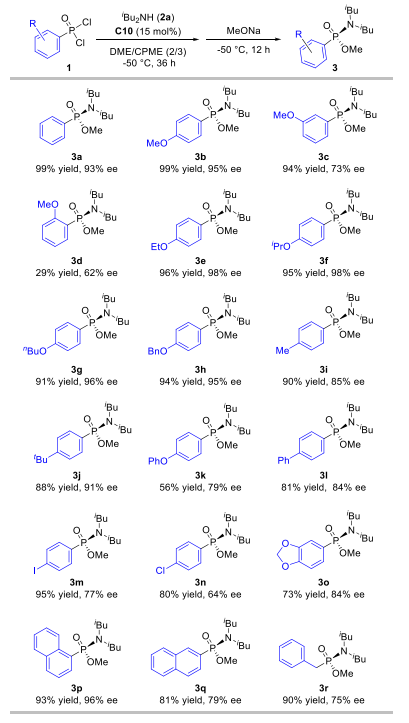
此后,作者系统考察了该催化体系对各类膦酰二氯底物的兼容性。研究发现苯环为对位带烷氧基的底物普遍能以高收率和高对映选择性获得目标产物(3b, 3e-3h);间位甲氧基取代底物延长反应时间至48小时后,产率为94%但选择性降低至73%(3c);而邻位取代底物则因空间位阻大、溶解性差,导致反应性明显下降(3d)。然而,当引入吸电子基团,反应的对映选择性会出现显著降低,延长第一步的反应时间至48小时,可以提高反应产率。该反应对二取代苯环底物(3o-3q)同样适用,并能兼容烷基膦酰二氯,例如苄基底物能以90%的产率和75%的ee值得到产物(3r)。这些结果一致表明,芳环上对位给电子取代基有助于提升磷中心的电子云密度,从而稳定催化中间体,实现更好的催化效果。作者还分离纯化了产物(3b)的前体化合物氯代膦酰胺,该化合物的对映选择性为96%。
总结
综上所述,上海交通大学张万斌团队使用课题组基于“键角调控”策略自主开发的手性双环咪唑催化剂,将前手性膦酰二氯的去对称化与立体专一的SN2亲核取代反应相结合,一锅法成功地构建了含C–P键的P(V)手性中心。该策略展现出优异的底物普适性与官能团兼容性,能以高产率和高对映选择性合成至少55种结构多样的手性膦氧化合物。机理研究证实,催化剂路易斯碱的亲核作用以及关键的N–H…O=P氢键作用的协同催化机制,是实现高对映选择性的核心。该方法的实用性在克级合成与药物分子(-)-SMT022332关键前体的制备中得到成功验证,为手性膦氧化合物的合成提供了一个强大平台。上述研究成果近期发表在J. Am. Chem. Soc.上,上海交通大学药学院博士生张露为论文的第一作者,化学化工学院张万斌教授和药学院张振锋研究员为论文的通讯作者。
文章详情:
本文摘抄自化学加公众号,仅用于学术传播,如有侵权请联系编辑部。
摘编:刘文燕


